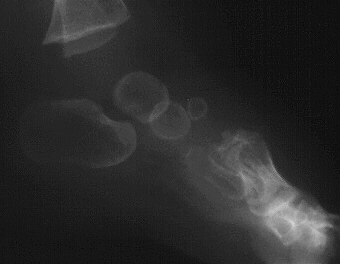
I was born September 16th, 1983, at 12:44am. My
mom had a smooth pregnancy, no complications. There was no indication that I was a dwarf prior to my birth, (I was my parents
first child). From what my parents were told, I was to be a healthy 6 lb baby. After I was born, they noticed something
was not "right". Therefore, I was rushed away. My parents were then told, that I would not live another hour (I'd LOVE to
meet the morons who told them that!). They later got a diagnosis, that I was a dwarf and had the
form called "Diastrophic Dysplasia" and I was put in the NICU (Neo-Natal Intensive Care Unit) for around 27 days until
my parents were allowed to take me home. My parents were put back, as in; they did not have a clue to what was going on. They
were then sent to genetic counselors. NEVERTHELESS, they handled everything VERY well! And I have a lot of respect for them!
They did not give up when they were told they should have. They went on to have my average height sister and then my diastrophic
sister Kathleen.
Diastrophic Dysplasia Info(Taken from the John Hopkins
website)
"Diastrophic dysplasia was first characterized by Maroteaux
and Lamy in 1960. Prior to this, patients with diastrophic dysplasia were described as having achondroplasia with clubfoot
or arthrogryposis multiplex congenita (a condition where a person has multiple joint contractures).
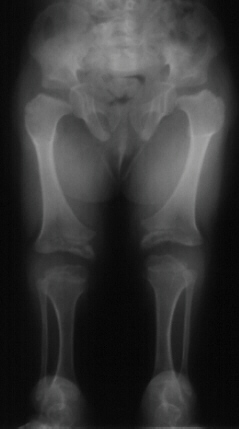
Physical features present at birth include short-limbed
dwarfism, hitchhiker thumb, and clubfeet. Abnormalities of the palate such as cleft palate or submucous cleft occur in 50%
of patients. The ears swell in the first days to weeks of life in 80% of individuals which then subsides spontaneously. Later,
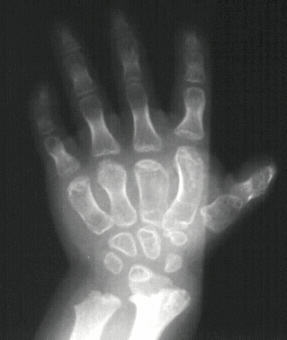
the ears have a cauliflower appearance. Fingers are short and broad with ulnar deviation. The thumb has a hitch-hiker
type appearance. There is increased mortality in infancy due to respiratory complications but thereafter, people with diastrophic
dysplasia have a normal life span.
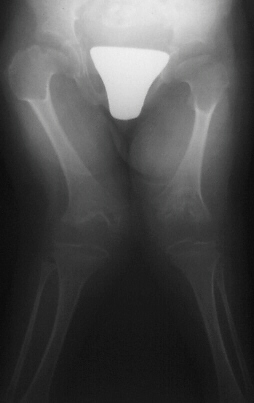
Orthopedic problems are common. The joints can be dislocated, especially
the shoulder, elbows, hips, and patellae (knee caps). Flexion contractures of knees and shoulders are common. Scoliosis is
not present at birth but often is progressive, especially in the early teens. Treatment of the scoliosis includes bracing
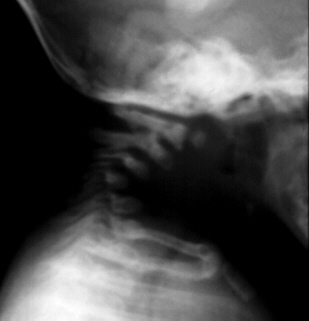
and occasionally, spinal fusion. Progressive cervical kyphosis can also occur with subluxation of the cervical spine which
can result in spinal cord compression.
The average length at birth is 42cms. Horton et. al. in
1982 published a growth curve of individuals with diastrophic dysplasia. The average adult height is 118cms with males ranging
from 86-127cms and females ranging from 104-122cms. Final height is influenced by the presence of scoliosis, hip and
knee contractures, and foot deformities.
Diastrophic dysplasia is inherited as an autosomal recessive condition.
This means that average-sized parents have a one in four (or 25%) chance of having additional children with diastrophic
dysplasia. Diastrophic dysplasia occurs at very low frequency in most populations but is seen frequently in Finland. The gene
for diastrophic dysplasia has been found and is called diastrophic dysplasia sulfate transporter (DTDST). Prenatal diagnosis
has been performed using ultrasound and by molecular DNA diagnosis."
My characteristics
OK, and the list goes............... sub-bloxed(sp) knees (the kneecaps were turned outwards), cauliflower ears, finger-joint fusion (the middle finger and ring finger on each hand, the
middle joint is fused), cleft pallet, and clubfoot. Those were visible at birth. At the age of 9 or so, I developed
scoliosis. And at age 11, I had a Spinal Fusion (which has held up since!). I had surgeries
to re-pair my knees, clubfeet (heel-cord lengthening, and cleft pallet. I am also starting to experience arthritis in my knees,
which from what I read, happens in 100% of cases.
Diastrophic dwarfism is a rare skeletal dysplasia first
defined by Maroteaux and Lamy in 1960. More than 200 cases have been described in literature (most from the U.S. and Finland).
The major clinical features of diastrophic dysplasia are:
severe short-limb short stature
cleft palate (27-59% of cases)
typical ear deformity (cauliflower deformity in 85% of
cases)
progressive deformities and contractures of joints (100%
of cases)
progressive hip dysplasia (dysplasia 70% of cases; dislocation
22% of cases)
typical hand deformities (100% of cases)
severe clubfoot (almost 100% of cases)
progressive spinal curvatures (Lumbar lordosis 100% of
cases, scoliosis 80% of cases)
Early degenerative changes in joints (100% of cases)
Genetic features:
Autosomic recessive transmission. ( D.T.D. and the McKusick
type metaphyseal chondrodysplasia are the only bone dysplasias with AR transmission).
D.T.D. gene has been located on chromosome 5. (That excludes
a primary defect of IX collagen, whose gene maps on chromosome 6)
5-6% of cases due to new mutations.
Prenatal diagnosis:
In the first trimester through DNA analysis
In the second trimester through US (short limb fetus with
abnormal metacarpophalangeal profile)
Diagnosis at birth can be suspected because of the typical
features.
Phenotypic variants:
1. Lethal form (death soon after birth because of cardio-
respiratory insufficiency)
2. Diastrophic variant (mild form with only some features)
Most patients have a normal life-span expectancy.
CLINICAL FINDINGS;
Average length at birth: 33 cm
Average adult height: 112 cm for both sexes (range 87
- 127 cm).
Short-limb dwarfism, usually rizomelic (40%) or mesomelic
(29%) .
Puberal growth spur does not occur in these patients.
Spinal deformities and hip and knee contractures accentuate the apparent dwarfism.
Nomocephalic head but typical facial appearance because
of the squared jaw, the narrow nasal bridge and the fullness of the circumoral area. These children are been called "cherub
dwarfs".
Cleft palate in 27 to 59% of cases (lower frequency in
diastrophic variants and higher in lethal variants).
Cauliflower ear deformity in 85% of typical DTDs and in
25% of diastrophic variants. It occurs during the first 6 weeks of life after an acute inflammatory process. Hearing impairment
not usual (it is related to fusion of ossicles).
Some patients present deformities of larynx and upper
airways (laryngo and tracheomalacia). They can develop severe respiratory insufficiency.
JOINTS:
The combination of marked limitation of motion of all major
joints together with a tendency to dislocation and subluxation characterize this disease. Some authors believe there are 2
forms of DTD: the lax and the stiff type.
Virtually every joint is likely to develop stiffness. This
is due to the severe deformities of bones (epiphyseal and metaphyseal) as well as soft tissue contractures.
Progressive dislocation of the hip, patella, radial head
are often observed.
Hip dislocation and hip dysplasia have
been reported, respectively in 22% and 70% of patients. Delayed femoral head appearing, coxa valga or, on the contrary, coxa
vara are common findings.
Valgus deformity of the knees, associated to flexion
contracture, is another common finding.
Clubfeet are another diagnostic feature
occurring in almost every patient. Clubfeet are usually very stiff and require surgical correction. Particular findings are
the adducted forefoot with a severe inward curvature of the metatarsals .
Hand deformities are essential for diagnosis
and they are present in almost 100% of cases. Hands are short and broad and deviated because of the ulnar shortness. PIP joint
stiffness is in contrasts with the hypermobility of the thumb that is abducted over a short first metacarpal ("hitch-hiker"
deformity).
Non-progressive lumbar lordosis is present in all
patients and it is probably related to flexion contractures of the hips. Cleft vertebral laminae are common in both cervical
and lumbar spine. Interpediculate narrowing occurs in 75% of patients but spinal stenosis is unusual because pedicles are
not short, the posterior arch is relatively normal.
Scoliosis or kyphoscoliosis occur in
80% of patients. These curves usually onset during the first 2 years of life and they are not due to primary vertebral deformities.
They must be carefully monitored because of the potential progression (usually during adolescence). Most authors suggest an
aggressive orthotic treatment and early spine fusion.
Diastrophic dwarfs do not present atlo-axial instability
or foramen magnum stenosis. In some cases their C-spine develops progressive kyphosis secondary to wedging of the lower
cervical vertebrae. Progression of this deformity can lead to neurologic deficits and death unless the patient undergoes posterior
or anterior and posterior spinal fusion.
The whole spine should be carefully monitored since
the first year of life.
RADIOGRAPHIC FINDINGS:
Spine: generally the vertebral bodies are normal before
the development of spinal deformities. As previously mentioned, cleft laminae are common. Deformities in C-spine vertebrae.
Long bones: they are broad and short. Metaphyses are flared
and expanded. A chevron-like shape is often present in femoral and tibial metaphyses. Epiphyseal centers appear late and are
severely irregular and flat. Ulna and fibula are usually short.
Tubular bones in hands and feet are short and broad with
typical deformities in first metacarpal and in metatarsals .
DIFFERENTIAL DIAGNOSIS:
Achondroplasia: no joint contractures (except elbows),
no clubfeet, hitchhiker thumb and ear deformities. Metaphyseal involvement (flared) but epiphyses are normal. Skull involvement.
Typical vertebral body deformities.
Arthrogryposis: no dwarfism, no epi-metaphseal involvement,
no ear deformity and hitchhiker thumb
SED: short-trunk dwarfism with stiff hips and often
cleft palate and clubfoot, but no thumb and ear involvement. Severe deformity of vertebral bodies and epi-metaphyseal involvement
of the proximal femur.
Larsen syndrome: typical flat face, multiple joint dislocations
present at birth. Sometimes cleft palate, clubfeet and progressive cervical spine kyphosis. No real dwarfism. No epi-metaphyseal
abnormalities and absence of the ear and thumb deformities.
TREATMENT:
Literature data are not enough to evaluate the orthopaedic
treatment of this disease.
Prevention and treatment of contractures, dislocations
as well as spinal and foot deformities should be the goal of the orthopedist.
Diastrophic dysplasia, which is also known as disastrophic
dwarfism, is a rare disorder that is present at birth (congenital). The range and severity of associated symptoms and physical
findings may vary greatly from case to case. However, the disorder is often characterized by short stature and unusually short
arms and legs (short-limbed dwarfism); abnormal development of bones (skeletal dysplasia) and joints (joint dysplasia) in
many areas of the body; progressive abnormal curvature of the spine (scoliosis and/or kyphosis); abnormal tissue changes of
the outer, visible portions of the ears (pinnae); and/or, in some cases, malformations of the head and facial (craniofacial)
area.
In most infants with diastrophic dysplasia, the first bone within the body of each hand
(first metacarpals) may be unusually small and "oval shaped," causing the thumbs to deviate away (abduction) from the body
("hitchhiker thumbs"). Other fingers may also be abnormally short (brachydactyly) and joints between certain bones of the
fingers (proximal interphalangeal joints) may become fused (symphalangism), causing limited flexion and restricted movement
of the finger joints. Affected infants also typically have severe foot deformities (talipes or "clubfeet") due to abnormal
deviation and fusion of certain bones within the body of each foot (metatarsals). In addition, many children with the disorder
experience limited extension, partial (subluxation) or complete dislocation, and/or permanent flexion and immobilization (contractures)
of certain joints.
In most infants with diastrophic dysplasia, there is also incomplete closure
of bones of the spinal column (spina bifida occulta) within the neck area and the upper portion of the back (lower cervical
and upper thoracic vertebrae). In addition, during the first year of life, some affected children may begin to develop progressive
abnormal sideways curvature of the spine (scoliosis). During adolescence, individuals with the disorder may also develop abnormal
front-to-back curvature of the spine (kyphosis), particularly affecting vertebrae within the neck area (cervical vertebrae).
In severe cases, progressive kyphosis may lead to difficulties breathing (respiratory distress). Some individuals may also
be prone to experiencing partial dislocation (subluxation) of joints between the central areas (bodies) of cervical vertebrae,
potentially resulting in spinal cord injury. Such injury may cause muscle weakness (paresis) or paralysis and/or life-threatening
complications.
In addition, most newborns with diastrophic dysplasia have or develop abnormal
fluid-filled sacs (cysts) within the outer, visible portions of the ears (pinnae). Within the first weeks of life, the pinnae
become swollen and inflamed and unusually firm, thick, and abnormal in shape. Over time, the abnormal areas of tissue (lesions)
may accumulate deposits of calcium salts (calcification) and eventually develop into bone (ossification). Some affected infants
may also have abnormalities of the head and facial (craniofacial) area including incomplete closure of the roof of the mouth
(cleft palate) and/or abnormal smallness of the jaws (micrognathia). In addition, in some affected infants, abnormalities
of supportive connective tissue (cartilage) within the windpipe (trachea), voice box (larynx), and certain air passages in
the lungs (bronchi) may result in collapse of these airways, causing life-threatening complications such as respiratory obstruction
and difficulties breathing. In some individuals with the disorder, additional symptoms and physical findings may also be present.
Diastrophic dysplasia is inherited as an autosomal recessive trait. .
Symptoms
The symptoms and physical findings associated with diastrophic
dysplasia may be extremely variable, differing in range and severity even among affected family members (kindreds). However,
in all individuals with the disorder, there is abnormal development of bones and joints of the body (skeletal and joint dysplasia).
During normal development before birth (embryonic and fetal development) as well as development
during early childhood, cartilage in many areas of the body is gradually replaced by bone (ossification). In addition, a layer
of cartilage (epiphyseal cartilage [growth plate]) separates the shafts (diaphyses) of long bones (e.g., bones of the arms
and legs) from their ends (epiphyses), allowing long bones to grow until the cartilage is no longer present. In those affected
by diastrophic dysplasia, however, there is delayed growth before and after birth (prenatal and postnatal growth retardation),
the development of the ends of the long bones (epiphyses) is irregular, and the ossification of the epiphyses is delayed.
Thus, affected newborns and children typically have markedly short, bowed arms and legs and short stature (short-limbed dwarfism).
In addition, in such cases, growth failure is typically progressive, in part due to absence of the "growth spurt" that usually
occurs during puberty. The severity of such growth failure may vary greatly from case to case, including among affected siblings.
Due to abnormalities of skeletal development, infants and children with diastrophic dysplasia also
have additional distinctive malformations of bones of the hands, feet, and other areas of the body. For example, the first
bone within the body of each hand (first metacarpals) may be unusually small, short, and "oval shaped." As a result, the thumbs
deviate away (abduction) from the body ("hitchhiker thumbs"). In addition, other fingers may be abnormally short (brachydactyly)
and joints between particular bones of the fingers (proximal interphalangeal joints) may become fused (symphalangism), causing
limited flexion and restricted movement (reduced mobility) of the finger joints. In some cases, bones of the wrists may also
be malformed due to premature ossification.
Infants with the disorder also typically have severe
foot deformities (talipes or "clubfeet") due to abnormal fusion and deviation of bones within the body of each foot (metatarsals).
In most cases, the heels turn outward (talipes valgus) while the fore part of each foot deviates inward (metatarsus adductus).
In other infants, the soles of the feet may be flexed (talipes equinus) and, in some cases, the heels may also turn inward
(talipes equinovarus). The great toes, like the thumbs, may also deviate away (abduction) from the body.
In addition to having limited flexion of finger joints, many affected infants and children also experience partial
dislocation (subluxation) and/or complete dislocation of particular joints of the body. For example, in many cases, dislocations
of the knees and hips occur upon weightbearing. Affected individuals may also have abnormally loose and/or stiff joints; experience
limited extension of joints at the elbows and/or knees; and/or develop permanent flexion and immobilization (contracture)
of certain joints (e.g., knees). Due to joint and bone abnormalities such as those affecting the feet, many individuals with
diastrophic dysplasia have a tendency to walk on tiptoe. In addition, affected individuals may be predisposed to degenerative
changes (osteoarthrosis) of particular joints (e.g. of the hips), resulting in pain with use of the joint, tenderness, stiffness,
and, in some cases, deformity.
Many infants with diastrophic dysplasia also have abnormalities
of bones within the spinal column (vertebrae). For example, in most affected infants, there may be incomplete closure of vertebrae
(spina bifida occulta) within the neck area and the upper portion of the back (lower cervical and upper thoracic vertebrae)
and/or abnormal narrowing of portions of the vertebrae of the lower back (interpedicular narrowing in lumbar vertebrae). During
the first year of life, some infants may begin to develop progressive abnormal sideways curvature of the spine (scoliosis).
In addition, during adolescence, individuals with diastrophic dysplasia may also develop abnormal front-to-back curvature
of the spine (kyphosis), particularly affecting vertebrae of the neck region (cervical vertebrae). In severe cases, progressive
kyphosis may result in difficulties breathing (respiratory distress). Some individuals with the disorder may also be prone
to experiencing partial dislocation of joints between the central areas (bodies) of cervical vertebrae (cervical subluxation),
potentially resulting in compression of the spinal cord. (This cylindrical structure of nerve tissue extends from the lower
portion of the brain and is located inside the central canal within the spinal column [spinal cavity].) Such spinal cord injury
may result in muscle weakness (paresis) or paralysis and/or life-threatening complications.
Most
newborns with diastrophic dysplasia also have or develop fluid-filled sacs (cysts) within the outer, visible portions of the
ears (pinnae). Within approximately two to five weeks after birth, the pinnae become swollen and inflamed. When such swelling
and inflammation subside, the pinnae remain unusually thick, hard, and abnormal in shape. The abnormal areas of tissue (lesions)
may gradually accumulate deposits of calcium salts (calcification) and eventually be replaced by bone (ossification). Although
affected infants may experience associated abnormal narrowing (stenosis) of the external ear canal (external auditory canal),
hearing is usually normal. However, according to reports in the literature, other affected infants and children may experience
hearing impairment due to such auditory canal stenosis or abnormal fusion or absence of the three tiny bones (auditory ossicles)
in the middle ear that conduct sound to the inner ear.
Some infants with diastrophic dysplasia
also have characteristic malformations of the head and facial (craniofacial) area, such as an unusually high, prominent forehead;
abnormal smallness of the jaws (micrognathia); and/or a broad, highly arched roof of the mouth (palate) or incomplete closure
of the palate (cleft palate). Cleft palate has been reported to occur in anywhere from 25 to 60% of affected infants, and
may cause difficulties with feeding and/or breathing. In addition, in some infants with diastrophic dysplasia, abnormalities
of supportive connective tissue (cartilage) within the windpipe (trachea), voice box (larynx), and air passages in the lungs
(bronchi) may cause abnormal narrowing (e.g., laryngotracheal stenosis) and collapse of such airways. In such cases, life-threatening
complications such as respiratory obstruction and difficulties breathing (respiratory distress) may result. However, in many
cases nasal speech (hyponasality) occurs as a result of the abnormally shaped vocal tract.
Approximately
one third of infants and children with diastrophic dysplasia also have dental abnormalities, such as abnormally small teeth
and dental crowding. In addition, in some cases, affected infants may have benign, reddish purple growths in the midportion
of the face (midline frontal hemangioma) due to an abnormal distribution of tiny blood vessels (capillaries). Some individuals
with the disorder may also have additional symptoms and physical findings. .
Causes
Diastrophic dysplasia is inherited as an autosomal recessive
trait. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received
from the father and one from the mother.
In recessive disorders, the condition does not appear
unless a person inherits the same defective gene for the same trait from each parent. If an individual receives one normal
copy of the gene and one mutated copy of the gene, the person will be a carrier for the disease but usually will not show
symptoms. The risk of transmitting the disease to the children of a couple, both of whom are carriers for a recessive disorder,
is 25 percent. Fifty percent of their children risk being carriers of the disease but generally will not show symptoms of
the disorder. Twenty-five percent of their children may receive both normal genes, one from each parent, and will be genetically
normal (for that particular trait). The risk is the same for each pregnancy.
Parents of some
individuals with diastrophic dysplasia have been closely related by blood (consanguineous). If both parents carry an altered
gene for the disorder, there is a higher than normal risk that their children may inherit the two genes necessary for the
development of the disease.
A gene responsible for diastrophic dysplasia, known as DTDST (for
"diastrophic dysplasia sulfate transporter" gene), has been located on the long arm (q) of chromosome 5 (5q32-q33.1). Chromosomes
are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes
are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females.
Each chromosome has a short arm designated as "p" and a long arm identified by the letter "q." Chromosomes are further subdivided
into bands that are numbered. For example, 5q32 refers to band 32 on the long arm of chromosome 5.
The
symptoms and findings associated with diastrophic dysplasia are thought to result due to abnormalities in the formation of
cartilage, thus affecting skeletal development. Early during normal embryonic development, the skeleton mainly consists of
cartilage that is gradually replaced by bone (ossification). After birth, many bones of the skeleton still consist primarily
of cartilage that will eventually ossify. However, researchers suspect that certain changes (mutations) of the DTDST gene
result in abnormalities of cartilage cells (chondrocytes) and the substance (matrix) that lies between such cells, ultimately
causing the symptoms and findings associated with the disorder. For example, in individuals with diastrophic dysplasia, the
growth plate of long bones may contain an abnormal distribution of cartilage cells (chondrocytes) and abnormal fibrous and
cystic areas within its matrix.
As discussed below (see "Affected Population"), diastrophic dysplasia
is particularly frequent in Finland. Genetic analysis has revealed that a specific mutation, designated as "DTDST(Fin)," is
present in affected members of many Finnish families (kindreds) and suggests that a single mutation event may have occurred
in a common ancestor (i.e., founder mutation) in the past. However, in some Finnish kindreds, the disorder has been shown
to result from different DTDST gene mutations (DTD-causing alleles) that do not descend from the common ancestral (founder)
mutation. In addition, different mutations of the DTDST gene have been identified in some non-Finnish individuals with the
disorder. .
Affected Populations
Diastrophic dysplasia affects males and females in equal
numbers. The disorder was originally recognized as a distinct disease entity based upon a 1960 report by investigators (Lamy
M, Maroteaux P) who discussed three observed cases as well as 11 similar cases previously recorded in the medical literature.
Since then, researchers have indicated that diastrophic dysplasia may be one of the most common forms of skeletal dysplasia.
Although the disorder appears to occur in most populations, it is thought to be particularly frequent in Finland, where reported
cases have included affected members in over 80 families (kindreds). Associated symptoms and physical findings may be extremely
variable from case to case, including among affected members of the same family. .
Related Disorders
Symptoms of the following disorders may be similar to those
of diastrophic dysplasia. Comparisons may be useful for a differential diagnosis:
Atelosteogenesis
type II, also known as neonatal osseous dysplasia I, is a rare genetic disorder caused by abnormal changes (mutations) of
the disease gene (DTDST) that is also responsible for diastrophic dysplasia (allelic disorder). Although the disorder has
many symptoms and findings similar to those associated with diastrophic dysplasia, it is typically more severe. Atelosteogenesis
type II is characterized by marked shortness of the arms and legs (micromelia), outward deviation (abduction) of the thumbs
and great toes, and severe deformity of the feet (talipes or "clubfeet") in which the soles are flexed and the heels are turned
inward (talipes equinovarus). Additional characteristic features include an unusually small chest (thorax), abnormal flatness
of certain bones in the spinal column (vertebrae), abnormal sideways curvature of the spine (scoliosis), front-to-back curvature
of vertebrae within the neck area of the spine (cervical kyphosis), and/or incomplete closure of the roof of the mouth (cleft
palate). Due to abnormalities of cartilage within the voice box (larynx), windpipe (trachea), and air passages in the lungs
(bronchi), affected infants may experience narrowing of the larynx (laryngeal stenosis), abnormal softness of cartilage in
the trachea and bronchi (tracheobronchomalacia), and underdevelopment of the lungs (pulmonary hypoplasia). Such abnormalities
may result in collapse of such airways, causing life-threatening complications shortly after birth, such as respiratory obstruction
and difficulties breathing (respiratory distress). Atelosteogenesis type II is inherited as an autosomal recessive trait.
Achondrogenesis type IB is a rare genetic disorder that is also thought to be caused by mutations
of the disease gene responsible for diastrophic dysplasia (allelic disorder). According to reports in the literature, the
disorder is more severe than diastrophic dysplasia and atelosteogenesis type II. Achondrogenesis type IB is characterized
by marked shortness of the arms and legs (micromelia) and short stature (short-limbed dwarfism), abnormally thin ribs, and
a susceptibility to rib fractures. Additional characteristic features include impaired ossification of vertebrae of the lower
back (lumbar vertebrae); the five fused bones forming the large triangular bone (sacrum) of the lower spine (sacral vertebrae);
and certain bones that form the hip bones (pubic and ischial bones). Achondrogenesis type IB is inherited as an autosomal
recessive trait.
Pseudodiastrophic dysplasia is a rare genetic disorder characterized by abnormally
short arms and legs and short stature (short-limbed dwarfism) and severe deformities of the feet (talipes or "clubfeet") that
tend to respond well to surgical treatment and physical therapy. Additional features may include dislocations of certain joints
in the fingers (proximal interphalangeal joints), dislocations of the elbows, flattening of the central regions of bones in
the spinal column (platyspondyly), abnormal sideways curvature of the spine (scoliosis), and/or other abnormalities. In contrast
to individuals with diastrophic dysplasia, the first bones within the body of each hand (first metacarpals) have a normal
appearance and the outer, visible portions of the ears (pinnae) do not experience the inflammation and cystic enlargement
often seen in those with diastrophic dysplasia in the first weeks of life. Pseudodiastrophic dysplasia is inherited as an
autosomal recessive trait.
Achondroplasia is a rare genetic disorder characterized by distinctive
abnormalities of the head and facial (craniofacial) area; unusually short upper arms and legs and short stature (short-limbed
dwarfism); and short hands with fingers that assume a "trident" or three-pronged position during extension. Affected individuals
may also have limited extension of the elbows and hips, bowing of the legs, and abnormally increased curvature of the bones
of the lower spine (lumbar lordosis). In addition, many individuals with achondroplasia have an abnormally enlarged brain
(macrencephaly), a prominent forehead (frontal bossing), and a flat (depressed) nasal bridge. In some cases, affected individuals
may experience inhibition of the normal flow of cerebrospinal fluid (CSF), potentially causing increased pressure on brain
tissue. In most cases, achondroplasia appears to occur randomly (sporadically) due to new genetic changes (mutations). In
other cases, the disorder may be inherited as an autosomal dominant trait. (For more information on this disorder, choose
"Achondroplasia" as your search term in the Rare Disease Database.)
Arthrogryposis multiplex
congenita is a group of disorders present at birth (congenital) that are characterized by limited movement or immobility of
several joints and partial or complete replacement of muscle with fibrous tissue in affected areas. Affected joints may be
permanently flexed or extended in various fixed postures (joint contractures). In many cases, the term arthrogryposis multiplex
congenita refers to a form of the disorder in which joint contractures result in abnormal extension of the elbows, flexion
of the wrists, and internal rotation of the shoulders. In addition, many affected individuals may have severe clubfoot (talipes
equinovarus), a deformity in which the heel is turned inward and the sole is flexed (plantar flexion). Additional associated
abnormalities may include a rounded face and a slightly small jaw. This form of the disorder appears to occur randomly (sporadically),
for unknown reasons. Another form of the disorder, known as a distal arthrogryposis (type 1), may be characterized by joint
contractures primarily affecting the hands and feet (distal limbs). Such contractures result in characteristic positioning
including permanent flexion (camptodactyly) and overlapping of fingers, deviation of fingers toward the "pinky" side of the
hand (ulnar deviation), clenching of the fists, and clubfeet. This form of the disorder is inherited as an autosomal dominant
trait. The causes of other forms of arthrogryposis multiplex congenita are variable. (For more information on this disorder,
choose "arthrogryposis multiplex congenita" as your search term in the Rare Disease Database.)
There
may be additional disorders that are characterized by growth delays before and after birth (prenatal and postnatal growth
retardation); abnormally short arms and legs and short stature (short-limbed dwarfism); distinctive malformations of bones
of the fingers and hands; clubfeet; partial (subluxation) or complete dislocation and/or permanent flexion and immobilization
(contractures) of certain joints; abnormal progressive curvature of the spine (e.g., scoliosis and/or kyphosis); and/or other
abnormalities similar to those potentially associated with diastrophic dysplasia. (For more information on such disorders,
choose the exact disease name in question as your search term in the Rare Disease Database.)
Standard Therapies
Diagnosis In some families with a previous
history of diastrophic dysplasia, it is possible that the disorder may be detected before birth (prenatally) during early
pregnancy (e.g., first trimester) based upon the results of specialized genetic (i.e., DNA marker) testing. In addition, in
some cases, the disorder may be detected during mid pregnancy (e.g., second trimester) through fetal ultrasonography, a specialized
imaging technique in which sound waves are used to create an image of the developing fetus. In such cases, diagnosis is most
easily established when a clear family history is present. During fetal ultrasonography, a diagnosis of diastrophic dysplasia
may be considered due to detection of certain characteristic findings, such as marked shortening of bones of the fingers (phalanges),
arms, and legs; abnormal deviation (abduction) of the thumbs ("hitchhiker thumbs") and great toes; severe deformities of both
feet (talipes or "clubfeet"); and/or other findings.
In most cases, diastrophic dysplasia is
diagnosed and/or confirmed at birth based upon a thorough clinical evaluation, identification of characteristic physical findings,
and a variety of specializing tests, such as advanced imaging techniques. For example, specialized x-ray studies such as computerized
tomography (CT) scanning and magnetic resonance imaging (MRI) may be used to detect, confirm, and/or characterize certain
skeletal abnormalities that may be associated with diastrophic dysplasia. During CT scanning, a computer and x-rays are used
to create a film showing cross-sectional images of internal structures. During MRI, a magnetic field and radio waves are used
to create cross-sectional images of organs and structures in the body.
Specialized diagnostic
testing (i.e., audiological tests) may also be performed to help detect hearing deficits that may occur in some children with
diastrophic dysplasia.
Treatment The treatment of diastrophic dysplasia is directed
toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team
of specialists who may need to work together to systematically and comprehensively plan an affected child's treatment. Such
specialists may include pediatricians; physicians who diagnose and treat abnormalities of the skeleton, joints, muscles, and
related tissues (orthopedists); surgeons; physical therapists; dental specialists (orthodontists); specialists who assess
and treat hearing problems (audiologists); and/or other health care professionals.
Specific therapies
for the treatment of diastrophic dysplasia are symptomatic and supportive. Physicians may carefully monitor affected infants
to ensure prompt detection and appropriate preventive or corrective treatment of respiratory obstruction and distress that
may result due to certain abnormalities potentially associated with the disorder (e.g., laryngotracheal stenosis). In addition,
special supportive measures may be used to help ensure an appropriate intake of nutrients in infants who experience feeding
difficulties due to cleft palate. In some cases, surgical procedures may be performed to correct malformations resulting in
breathing and/or feeding difficulties. The specific procedures performed will depend upon the location, severity, and combination
of such anatomical abnormalities.
In addition, various orthopedic techniques, including surgery,
may also be used to help prevent, treat, and/or correct certain skeletal deformities associated with diastrophic dysplasia.
In some cases, physical therapy in combination with surgical and supportive measures may be helpful in improving an affected
individual's ability to walk and perform other movements (mobility). According to the medical literature, although the foot
deformities (i.e., talipes or clubfeet) associated with the disorder may be resistant to treatment, early, persistent therapy
may be helpful in achieving beneficial results. In addition, because particular skeletal changes associated with diastrophic
dysplasia are progressive (e.g., kyphosis) and, in some cases, may lead to severe complications (e.g., respiratory distress,
compression of the spine, potential paresis or paralysis), physicians may perform ongoing monitoring to ensure prompt detection
of and appropriate preventive and/or corrective measures for such abnormalities.
In affected
children with dental abnormalities, braces (orthodontics), dental surgery, and/or other corrective procedures may be undertaken
to correct such malformations. Steroid injections and/or other measures may also be used to help decrease the ear deformity
that often affects infants with the disorder.
Genetic counseling will be of benefit for affected
individuals and their families. Other treatment for this disorder is symptomatic and supportive.